A 24-year-old graduate student is brought to the ED after calling 911 stating that she overdosed on her antidepressant medication. She is alert, crying, and complains of a dry mouth. Her initial heart rate is 96 bpm with a blood pressure of 120/80 mmHg. Her examination is remarkable for dilated pupils, flushed skin, and occasional twitching of muscles in her arms and legs. The baseline ECG demonstrates sinus rhythm with a markedly prolonged QT interval. The patient suddenly becomes lethargic and a 12-lead ECG prompts the emergency physician to consider urgent therapy.

Torsade de pointes
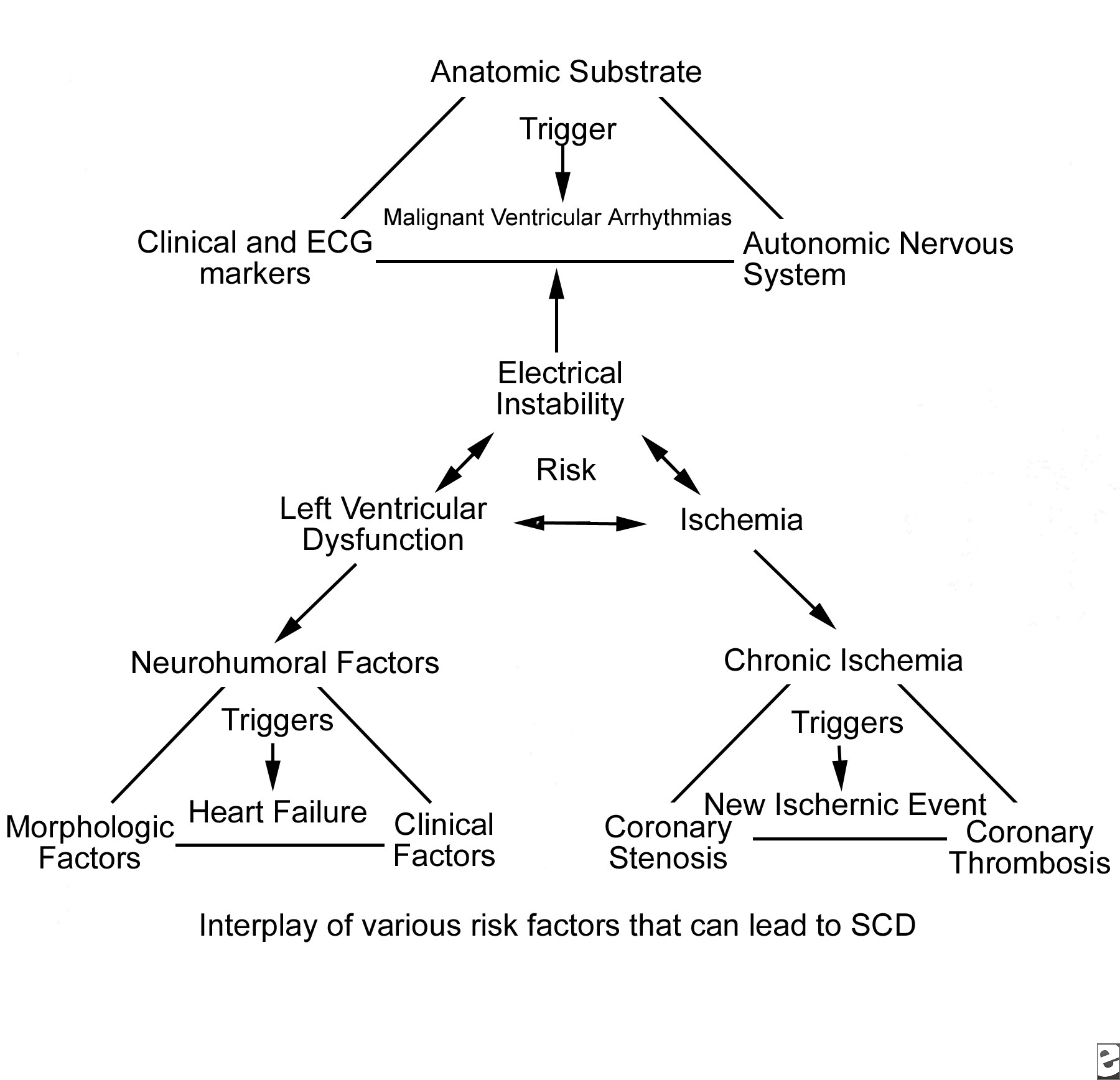
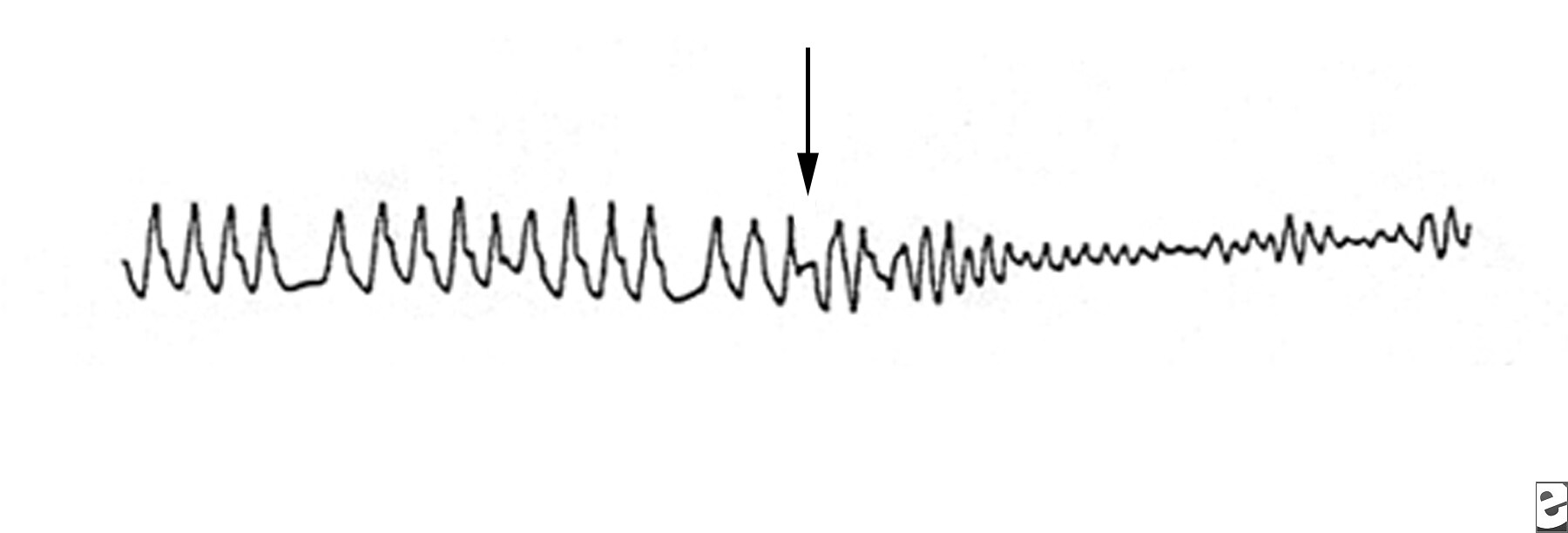
Torsade de pointesTorsade de pointes is a syndrome of ventricular tachycardia in which the electrical axis “twists” around. The QRS complex exhibits a crescendo-decrescendo variation in amplitude. The R-R interval is frequently in the range of 200–250 bpm. One of the characteristic features of torsade is a long period of ventricular repolarization so that the QT interval is typically at least 500 ms long. This prolonged QT interval is most readily observed in the QT interval immediately prior to the onset of torsade. Most cases of torsade are preceded by long-short R-R cycles. For example, after a premature ventricular complex a compensatory pause will occur, and then a sinus beat with a long QT interval will occur. If another PVC occurs, torsade may be initiated. If a premature stimulus occurs near the zenith of the T wave, it may be more likely to induce a ventricular arrhythmia. However, a short couple variant with a particularly high mortality has been described. It is important to distinguish polymorphic VT with a normal QT interval from torsade as the treatment and prognosis may be different.
The QT interval is measured from the onset of the Q wave to the end of the T wave. The QT interval can vary with heart rate. Bradycardia is often associated with a prolonged QT interval while tachycardia is associated with a shortened QT interval. The QT interval can be corrected (QTc) for heart rate using Bazett’s formula (the QTc equals the longest QT interval divided by the square root of the preceding R-R interval). If atrial fibrillation is present, the QTc should be measured for 10 consecutive beats and averaged. Correct assessment of the QTc is critical during initiation of sotalol in patients with atrial fibrillation.
Torsade may devolve into ventricular fibrillation, return to the baseline rhythm, or end with asystole. Therefore, the first line of therapy is usually defibrillation followed by intravenous magnesium sulfate. Once the patient has hemodynamics that allow perfusion of vital organs, the goal is identifying the underlying cause. Common causes include extreme bradycardia, congenital causes of long QT syndrome, anti-arrhythmic drugs, and one or more combinations of drugs that prolong the QT interval. Both drug overdose and reduced drug clearance can prolong the QTc sufficiently to cause torsade.In a retrospective study of 249 cases of torsade not attributed to cardiac drugs, Zeltser and colleagues noted that 71% of the cases involved female patients. Other risk factors in this series included hypokalemia, the use of multiple drugs that prolong the QT interval, increased drug dosage, a history of prior torsades, and a family history of long QT syndrome. Among cardiac medications, the class IA and class III anti-arrhythmics are associated with the development of torsade. The class IA drugs are most well known for blocking sodium channels. However, at low serum drug concentrations, potassium ion current blockage occurs. The association of quinidine is well described in the literature. Disopyramide has also been implicated. Nacteylprocainamide, a metabolite or procainamide, can cause torsade via QT prolongation by blocking the Ikr channel. Class III anti-arrthymics are potent Ikr channel-blockers. High serum concentrations of these drugs, either due to overdose or decreased clearance, can result in torsade. As these drugs exhibit reverse use dependence, Ikr is more effectively blocked at slow heart rates. Thus, bradycardia increases the risk of torsade with class III agents. Interestingly, the class III agent amiodarone is rarely associated with torsade. Drouin and colleagues [25] demonstrated that amiodarone decreases heterogeneous repolarization, thus reducing the susceptibility of re-entry. Torsade is also associated with overdose of tricyclic anti-depressants and with use of the neuroleptins, including phenothiazines and haloperidol. Among antimicrobials, the macrolides erythromycin and clarithromycin have been reported to prolong the QT interval and cause torsade. Both of these medications inhibit the CYP3A4 system. Therefore, QT prolongation may occur in a patient taking either of these antibiotics with another drug that is metabolized by the CYP3A4 system. Such drugs will cause QT prolongation with increasing serum concentrations. The incidence of torsade among patients taking azithromycin is substantially less than that of patients taking erythromycin. An interesting historical footnote is cisapride, a promotility drug withdrawn from the U.S. market because it has a high incidence of QT prolongation and arrhythmia. Cisapride blocks the Ikr channel. Finally, in the case of bradycardia with long QT, temporary pacing may be necessary to prevent recurrence until the etiology of the slow heart rate can be diagnosed and treated.
Critical Decisions in Emergency and Acute Care Electrocardiography.
Edited by William J. Brady and Jonathon D. Truwit © 2009 Blackwell
Critical Decisions in Emergency and Acute Care Electrocardiography.
Edited by William J. Brady and Jonathon D. Truwit © 2009 Blackwell